- 1️⃣ 先天性高胰島素血症(Congenital hyperinsulinism, CHI)
- 2️⃣ 全腦下垂體功能低下症(Panhypopituitarism)
這兩個若 rule out,基本上就要思考內分泌以外的其他原因(例如先天性代謝異常、用藥⋯等)。
臨床上做好下面三件事,就可以在照會時加快鑑別診斷的效率 👇
病史詢問(History taking)
以下族群都可能造成 neonatal hypoglycemia:
- 1️⃣ 早產兒 2️⃣ 過熟兒 3️⃣ SGA 4️⃣ LGA 5️⃣ GDM(妊娠糖尿病母親) 6️⃣ Low Apgar score 7️⃣ Perinatal asphyxia
(便當沒帶夠就跑出來上學)
1️⃣ 早產兒 · 3️⃣ SGA
(胰島素分泌太多,又沒辦法精準調控)
2️⃣ 過熟兒 · 4️⃣ LGA · 5️⃣ GDM · 6️⃣ Low Apgar · 7️⃣ Asphyxia
IUGR / asphyxia 相關者可能持續數月;以上族群同時也是新生兒科考慮 high risk for hypoglycemia 的對象(第一天積極驗血糖:1、4、6、6、6 hr)。
理學檢查(Physical exam)
因 insulin 本身即 growth factor,通常一出生就可看見顯著的肥胖寶寶(macrosomia)。
因 counter-regulatory hormone(GH、ACTH)不足,除反覆低血糖(可能嚴重到以 seizure 表現)外,可仔細留意以下線索(hint):
- 1️⃣ TSH 不足 → 續發性甲狀腺低下:widen fontanelle、dry skin、umbilical hernia、prolonged jaundice(indirect)、macroglossia
- 2️⃣ FSH/LH 不足 → hypogonadotropic hypogonadism:micropenis、cryptorchidism
- 3️⃣ ACTH 不足 → 續發性腎上腺功能不全:prolonged jaundice(direct)、hypotension + hyponatremia(salt-wasting crisis)、fluid retention
有些類型可發現 hepatomegaly。
若為原發性(1st)腎上腺功能不全,除上述特徵外,還會多出現 hyperpigmentation(ACTH 分泌時順帶產生 MSH)與 hyperkalemia(aldosterone 一起不夠)。
Critical sampling
因 endocrine response 不一定即時,若病人沒有 life-threatening S/S,請把 0 分鐘與 30 分鐘檢體留滿後再補 glucose water:
- Blood gas
- Central glucose · One-touch
- GH · Cortisol · Insulin
- Ketone
- NH3 *
- Lactic acid
- Tandem mass analysis *
- Urine organic acid analysis(GC/MS)*
- Plasma amino acid *
兩個提醒:①急診病人、有暴露降血糖藥(含注射型胰島素)疑慮 → 加抽 0 與 30 分鐘 C-peptide。②留檢體有困難時,打 * 的項目(NH3、Tandem mass、Plasma amino acid、Urine)可在處理完低血糖後再行留檢。
基本生理學(Physiology of blood glucose control)
- Insulin:「降」血糖、「抑制」肝醣分解(Glycogenolysis)與糖質新生(Gluconeogenesis)、「促進」血糖進入細胞使用(GLUT 1–5、SGLT 1–2)。
- Counter-regulatory hormone:「升」血糖、「促進」肝醣分解與糖質新生、「抑制」血糖進入細胞使用(GLUT 1–5、SGLT 1–2)。包含 👉 Glucagon、Catecholamines、GH、Cortisol。
| Hormone | 血糖效果 | 機轉簡述 |
|---|---|---|
| Glucagon | 升糖 ↑ | 主要作用於肝臟,快速促進肝醣分解與糖質新生(升糖第一線) |
| Catecholamines 兒茶酚胺 | 升糖 ↑ | 促進肝醣分解、糖質新生與脂解;抑制胰島素分泌、降低周邊葡萄糖利用 |
| Growth hormone GH | 升糖 ↑ | 降低周邊組織葡萄糖利用、促進脂解(作用較慢,屬長期反調節) |
| Cortisol | 升糖 ↑ | 促進糖質新生、增加受質供應,對其他升糖激素具 permissive 作用(作用較慢) |
*各荷爾蒙皆為「升糖」;欄位「機轉簡述」為常見教科書要點整理,可依需要增刪。
胰島素(左)與反調節荷爾蒙(右)對五個代謝過程作用方向相反,共同決定血糖。(依原圖以程式碼重繪)
Brook's Clinical Pediatric Endocrinology, 7th ed., Chapter 16.
胰島素分泌(Insulin secretion)
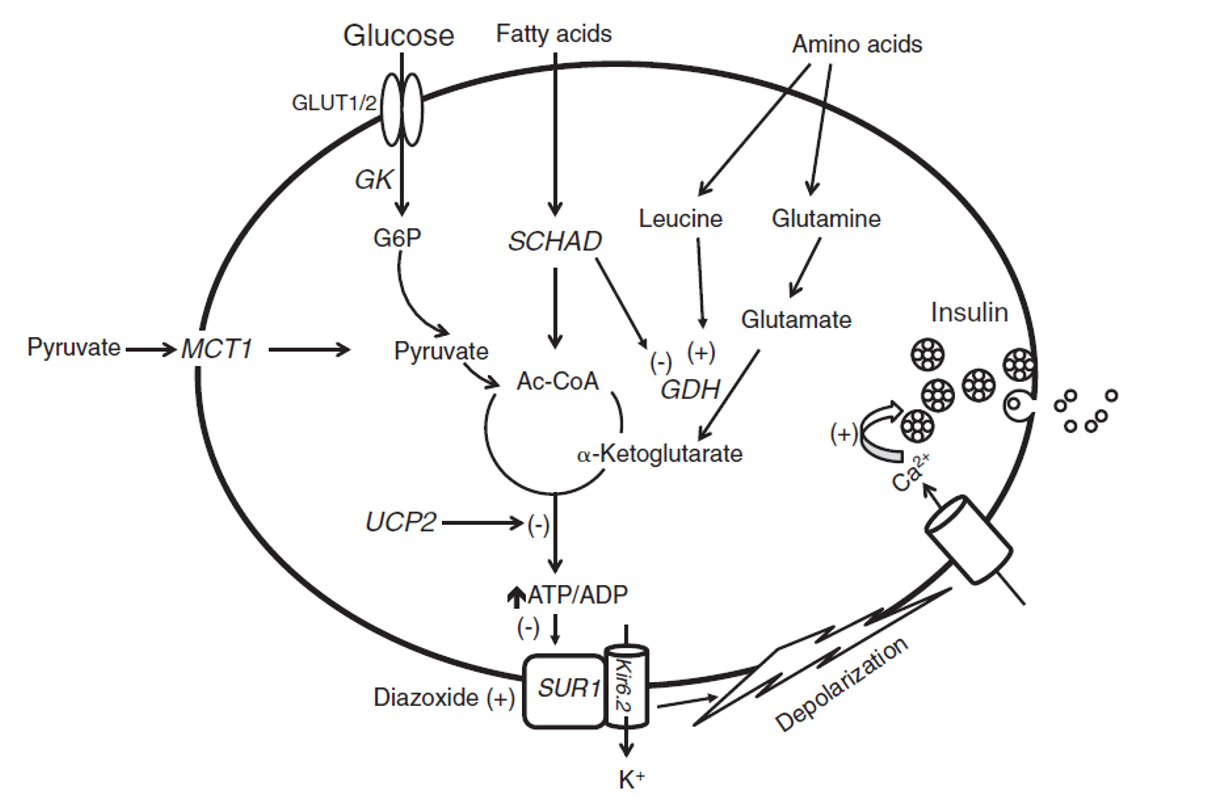
胰臟 β 細胞葡萄糖感應與胰島素分泌路徑(GLUT → GK → ATP/ADP↑ → KATP 關閉 → 去極化 → Ca²⁺ → 胰島素釋放)。
Brook's Clinical Pediatric Endocrinology, 7th ed., Chapter 16.
- 1️⃣ 血糖透過 Glucose Transporter(GLUT) 進入胰臟 β 細胞。
- 2️⃣ 進入後經一系列酵素作用產生 ATP。
- 3️⃣ ATP/ADP ratio 提高 → KATP channel「關閉」。
- 4️⃣ K⁺ 累積於細胞內造成去極化,Ca²⁺ 通道打開 → 促進胰島素釋放。
低血糖的臨床症狀(Signs & symptoms)
血糖快速下降時、由反調節反應引起,常是較早期的警示:
- 😰 冒冷汗、發抖、心悸/心跳快
- 😨 焦慮不安、臉色蒼白
- 🍽️ 飢餓感、噁心
腦部葡萄糖不足所致,較嚴重、需盡快矯正:
- 🧠 意識改變、注意力不集中、行為異常
- 👁️ 視力模糊、頭痛、嗜睡
- ⚠️ 抽搐、昏迷
躁動顫抖、餵食差、低張力、體溫過低、呼吸暫停/發紺、高音哭聲、抽搐;症狀常不專一,需主動監測血糖而非等症狀出現。
低血糖的定義(Definitions of hypoglycemia)
具備以下三者,才算「有臨床意義」的低血糖:
- 1️⃣ 有符合低血糖的症狀/徵象。
- 2️⃣ 症狀出現當下,測得血糖偏低(建議以實驗室血漿葡萄糖確認)。
- 3️⃣ 給予葡萄糖、血糖回升後,症狀隨之緩解。
新生兒/嬰兒症狀常不典型(如躁動、餵食差、低張力、抽搐),故三要件不一定齊全,需搭配下方切點判讀。
兩套隨出生時間變動的低血糖切點對照(依原圖以程式碼重繪)。
Pediatric Endocrine Society (PES) consensus workshop.
台大兒童醫院住院醫師工作手冊,NB 章節。
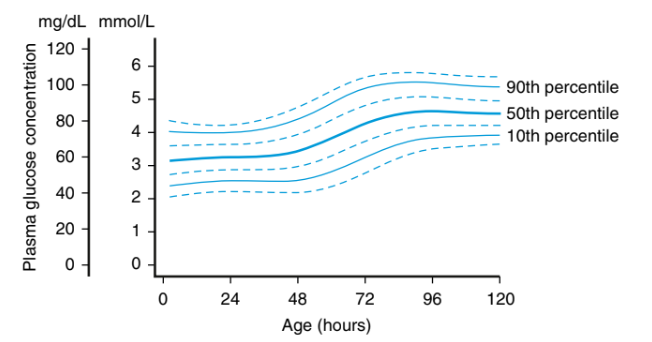
出生後 0–120 小時血漿葡萄糖濃度百分位(10th / 50th / 90th)。
Nelson Textbook of Pediatrics, 21st ed., Chapter 113.
- 1️⃣ 出生後 4 hr:40 mg/dL
- 2️⃣ 出生後 4–24 hr:45 mg/dL
- 3️⃣ 出生後 24–48 hr:50 mg/dL
- 4️⃣ 出生後 48–72 hr:60 mg/dL
- 5️⃣ 出生後 ≥ 72 hr:70 mg/dL
靜脈葡萄糖矯正(IV glucose correction)
- 👉 2 mL/kg D10W(0.2 g/kg = 200 mg/kg)
- 👉 下 Order 的方式「2 mL/kg D10W IV push」
- ⚠️ 以上做法可降低血糖 rebound、同時避免滲透壓改變太大造成 IVH 風險
- ⚠️ bolus 後接續 GIR 維持(嬰兒約 5–8 mg/kg/min),目標血糖 > 70 mg/dL
Thornton PS, Stanley CA, De Leon DD, et al.; Pediatric Endocrine Society. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015;167(2):238–245. doi:10.1016/j.jpeds.2015.03.057
Adamkin DH; AAP Committee on Fetus and Newborn. Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics. 2011;127(3):575–579. doi:10.1542/peds.2010-3851
🙏 特別感謝台大新生兒科 黃信中醫師 協助校稿討論並給予實務建議。
- 👉 5 mL/kg D10W = 1 mL/kg D50W 稀釋成 4 mL/kg(0.5 g/kg = 500 mg/kg)
- 👉 下 Order 的方式(統一以 D50W 為例):「1 mL/kg D50W 稀釋成 4 mL/kg, IV push」
- ⚠️ 在兒科急救中,為了達到 0.5 g/kg 的目標給糖量,有一個著名的「50 法則」(濃度 × 體積 = 50)
📖 Ref:American Heart Association (AHA). Pediatric Advanced Life Support (PALS) Provider Manual.
- 👉 2 mL/kg D25W = 1 mL/kg D50W 稀釋成 2 mL/kg(0.5 g/kg = 500 mg/kg)
- 👉 下 Order 的方式(統一以 D50W 為例):「1 mL/kg D50W 稀釋成 2 mL/kg, IV push」
- ⚠️ 在兒科急救中,為了達到 0.5 g/kg 的目標給糖量,有一個著名的「50 法則」(濃度 × 體積 = 50)
📖 Ref:American Heart Association (AHA). Pediatric Advanced Life Support (PALS) Provider Manual.
- 👉 1 mL/kg D50W(0.5 g/kg = 500 mg/kg)
- 👉 下 Order 的方式(統一以 D50W 為例):「1 mL/kg D50W 不用稀釋, IV push」
- ⚠️ 在兒科急救中,為了達到 0.5 g/kg 的目標給糖量,有一個著名的「50 法則」(濃度 × 體積 = 50)
- ⚠️ 實務上限補充(Optional):雖然 1 mL/kg 是準確的法則,但在成人急診實務上,推注的上限通常就是直接給予 2–3 支 D50W(1 Ampule = 20 mL)
📖 Ref:American Heart Association (AHA). Pediatric Advanced Life Support (PALS) Provider Manual.
若無 life-threatening 症狀,記得先留 critical sample 再補糖(見「臨床 Approach」章節)。
Critical sampling
血糖驗到 ≤ 50 mg/dL 時抽好抽滿;endocrine response 不一定即時,若無 life-threatening S/S,0 與 30 分鐘檢體留滿後再補 glucose water:
- Blood gas
- Central glucose · One-touch
- GH · Cortisol · Insulin
- Ketone
- NH3 *
- Lactic acid
- Tandem mass analysis *
- Urine organic acid analysis(GC/MS)*
- Plasma amino acid *
- 如病人沒有 life-threatening S/S,請留到 critical sample 後再補 glucose water!
- 急診病人、有暴露降血糖藥(含注射型胰島素)疑慮 → 加抽 0 與 30 分鐘 C-peptide。
- 留檢體有困難時,打 * 的項目(NH3、Tandem mass、Plasma amino acid、Urine)可在處理完低血糖後再行留檢。
DD 流程圖(依 critical sample 分流)
以 critical sample(HCO₃、Lactate、BOHB、FFA)分流之低血糖鑑別診斷(依原圖以程式碼重繪)。
高胰島素血症(Hyperinsulinaemic hypoglycemia)
胰島 β 細胞胰島素分泌失調,是新生兒持續性低血糖最常見原因。
- Hypoketotic + hypofattyacidaemic hypoglycemia(胰島素抑制脂解與酮生成)
- Insulin / C-peptide / proinsulin 不當偏高
- 腦部同時失去主要(glucose)與次要(ketone)燃料 → 腦傷風險最高
| Parameter(血糖 < 50 mg/dL 時抽驗) | Sensitivity (%) | Specificity (%) |
|---|---|---|
| Insulin > 2 μU/mL a | 82.2 | 100 |
| C-Peptide ≥ 0.5 ng/mL | 88.5 | 100 |
| β-hydroxybutyrate < 1.8 mmol/L | 100 | 100 |
| Free Fatty Acids < 1.7 mmol/L | 87 | 100 |
| IGFBP-1 ≤ 110 ng/mL | 85 | 96.6 |
| Glycemic response to glucagon > 30 mg/dL | 89 | 100 |
a 依 insulin assay 敏感度而定。IGFBP=Insulin-like growth factor binding-protein。(Sperling Table 7.3,以程式碼重繪)
🎯 病因分類- 1️⃣ 早產兒
- 2️⃣ 過熟兒
- 3️⃣ SGA
- 4️⃣ LGA
- 5️⃣ GDM(妊娠糖尿病母親)
- 6️⃣ Low Apgar score
- 7️⃣ Perinatal asphyxia
主要致病基因如下表。下表所列 gene 皆為胰島素分泌過程受阻導致的 monogenic CHI(可搭配「先備知識」的胰島素分泌圖較好理解),最常見為 ABCC8/KCNJ11 mutation,又可分成:
- 👉 Diazoxide unresponsive:ABCC8/KCNJ11、GCK
- 👉 Diazoxide responsive:GLUD1、HADH(SCHAD)、HNF4A、HNF1A、SLC16A1(MCT1)、UCP2、FOXA2
| 基因 | 重點 |
|---|---|
| ABCC8 / KCNJ11 | 🧬 Inactivating function mutation 造成 KATP channel 無法打開
🧬 Autosomal dominant(較輕微)or recessive(較嚴重)
👉 CHI 中最常見與最嚴重的 subtype,又可依對胰臟的影響分成 diffuse type 與 focal type
👉 Diffuse type:發生在 germline 的 autosomal recessive mutation 最常見
👉 Focal type 的 2-hit mechanism:
1️⃣ 發生在 Germline:paternal(父源) mutation of the ABCC8 or KCNJ11 gene
2️⃣ 發生在 Somatic(胰臟):maternal(母源) deletion of the ABCC8 or KCNJ11 gene
⚠️ 若血液 genetic study 只找到一股(父源)的 ABCC8 或 KCNJ11 recessive mutation,則要特別注意可能是 focal type;開刀時要多留胰臟檢體做 genetic study 確認有無 somatic deletion
|
| GCK | 🧬 Gain-of-function mutation 造成 glucose 進入 β cell 的 threshold 降低
🧬 Autosomal dominant
🧬 如果是 inactivating mutation:
1️⃣ Heterozygous(MODY2)
2️⃣ Homozygous(Neonatal diabetes)
|
| GLUD1 | 🧬 Gain-of-function mutations
🧬 Autosomal dominant
👉 CHI 中第二常見的 subtype,diazoxide responsive 中最常見的 subtype
👉 會被 fasting 與 protein-rich meals 誘發
👉 Ammonia 也會異常升高(正常值 3–5 倍以上,約 60–150 μmol/L),故又稱 The Hyperinsulinism Hyperammonemia Syndrome(HI/HA)
👉 Seizures、learning disability、behavioral disorders 等發生頻率增高,且似乎與低血糖無直接相關
|
| HADH (SCHAD) | 🧬 Inactivating mutations 造成 GDH 的調節出問題
🧬 Autosomal recessive
👉 機轉:SCHAD(粒線體 short-chain 3-hydroxyacyl-CoA dehydrogenase)在 β 細胞中扮演「抑制 GDH 活性」的角色;SCHAD 缺損 → 失去這層抑制 → GDH 過度活化 → 胰島素分泌失調(故臨床表現與 GDH-HI 相似)
👉 與 GDH-HI 同樣會有 protein-induced hypoglycemia;但不會有 ammonia 升高(SCHAD 在 GDH 表現的其他組織中表現量較低)
👉 與其他 fatty acid oxidation defects 不同:無肝功能異常、心肌病變或骨骼肌影響
👉 生化指標:血漿 3-hydroxybutyryl-carnitine ↑、尿液 3-hydroxyglutarate ↑
👉 Diazoxide 有反應;表現異質性大,從晚發輕症到新生兒期嚴重低血糖皆有
|
| HNF4A | 🧬 Inactivating mutations
🧬 Autosomal dominant
1️⃣ 早期:persistent hypoglycemia caused by hyperinsulinism
2️⃣ 青少年/early adult:reduced insulin secretion 造成 MODY1
|
| HNF1A | 🧬 Inactivating mutations
🧬 Autosomal dominant
1️⃣ 早期:persistent hypoglycemia caused by hyperinsulinism
2️⃣ 青少年/early adult:reduced insulin secretion 造成 MODY3
|
| SLC16A1 (MCT1) | 🧬 Inactivating mutations
🧬 Autosomal dominant
👉 運動誘發高胰島素(EIHI),避免劇烈運動
|
| UCP2 | 🧬 Inactivating mutations
🧬 Autosomal dominant
|
| FOXA2 | 🧬 Inactivating mutations
👉 較新發現的型別,特色是 HI 合併 hypopituitarism(腦下垂體功能低下)
👉 機轉:Foxa2 為轉錄因子,參與多種組織(含腦下垂體)的發育,同時在成熟 β 細胞的胰島素分泌中扮演重要角色
👉 表現:新生兒期 hypoketotic hypoglycemia
👉 治療:對腦下垂體荷爾蒙補充與 diazoxide 皆有反應
⚠️ 提醒:新生兒期 hypopituitarism 的低血糖可以「長得像」HI(此時期 ketogenesis 機制尚未成熟);一般 hypopituitarism 對 diazoxide 無反應、需補充缺乏的荷爾蒙才會緩解——但 FOXA2 是兩者兼具的例外
|
CHI 致病基因(以程式碼重繪)。
Sperling Pediatric Endocrinology, 6th ed., Chapter 7.
部分症候群本身即會合併高胰島素低血糖,多為暫時性但少數會持續,需個別評估:
| Syndrome(症候群) | 重點 |
|---|---|
| Beckwith-Wiedemann syndrome (BWS) | 🧬 Chromosome 11p15.5 imprinting disorder——過度生長(overgrowth)+腫瘤易感症候群,源自兩個 imprinted control center 的遺傳/表觀遺傳變化
• Domain 1(遠端):IGF2(促生長,父源表現)、H19(lncRNA 生長抑制,母源表現)
• Domain 2(近端):KCNQ1OT1(父源)、CDKN1C(母源,細胞增生負調控)、KCNQ1(母源)
🧬 成因比例:IC2 甲基化缺失 50%/父源單親等二倍體 20%/IC1 甲基化增加 5%/母源 CDKN1C 突變 5%
👉 Clinical features:
• Macrosomia(過度生長)
• Macroglossia(巨舌)
• Visceromegaly(內臟腫大)
• Abdominal wall defects(腹壁缺損)
• Ear creases/pits(耳摺痕/耳凹)
• Body asymmetry(身體不對稱)
• 胚胎性腫瘤風險增加(embryonal tumor)
👉 高胰島素低血糖發生於高達 50% 的個案;機轉未明,可能涉及胰島素分泌失調與 β 細胞增生
👉 治療:多為輕微且暫時性,部分嚴重且持續。部分對 diazoxide 有反應,少數需胰切除;因多數在一歲內緩解,胰切除應保留給藥物無法控制者
|
| Kabuki syndrome | 👉 syndromic hyperinsulinism 中第二常見者
🧬 70–75%:KMT2D(lysine-specific methyltransferase 2D)mutations,autosomal dominant(多為 de novo)
🧬 1–9%:X-linked KDM6A(lysine-specific demethylase 6A)mutations
👉 Clinical features(cardinal features):
• 典型「Kabuki Mask」臉部特徵:long palpebral fissure、arched eyebrows、下眼瞼外側 1/3 外翻
• Skeletal abnormalities(骨骼異常)
• Dermatoglyphic abnormalities(皮紋異常)
• Neurodevelopmental delay、intellectual disabilities
• Postnatal growth retardation(出生後生長遲滯)
👉 Endocrine manifestations:
• 22% GH deficiency
• 20% Premature thelarche
• 11% Neonatal hypoglycemia
• 10% Hypothyroidism
👉 新生兒低血糖的機轉可能是多因性:hyperinsulinism + GH deficiency
👉 Hyperinsulinism 見於高達 6% 的 Kabuki 個案;通常出生即存在、diazoxide 有反應,多在十歲前緩解
|
| Turner syndrome | 🧬 機轉未明,X 染色體上的候選基因為 KDM6A(與 Kabuki syndrome 相同的 histone demethylase)
👉 Turner 除了已知易罹患 T1DM/T2DM 外,也易發生低血糖(較少被認識);低血糖個案中 Turner 的比例高於預期
👉 多在出生後不久表現,通常對 diazoxide 有反應;少數嚴重且無反應者需胰切除
👉 病理:islet cell nucleomegaly,與 KATP inactivating mutation 造成的 diffuse HI 相似;胰島功能顯示基礎胞內鈣與胰島素上升、GSIS threshold 下降、對胺基酸刺激敏感度增加,對 glyburide 反應保留
|
| Other syndrome | Sotos syndrome、Simpson-Golabi-Behmel syndrome、Perlman syndrome、Costello syndrome⋯⋯
|
Sperling Pediatric Endocrinology, 6th ed., Chapter 7.
- 目標:血糖 ≥ 70 mg/dL
- 方法(依優先次序由上往下):
- 1️⃣ IV 糖水:SIR(糖輸注速率)可能需拉到正常值 5 倍以上。
- 2️⃣ Glucagon
• Bolus:0.5–1 mg 或 20–30 μg/kg(可持續 40–60 min,未建立 IV 時爭取時間)
• Pump:2.5–20 μg/kg/h - 3️⃣ Diazoxide:5–15 mg/kg/day,Q12H–Q8H
⚠️ 須至少使用五天才能判斷有無 responsive
⚠️ 擔心 fluid retention,同步使用利尿劑:Chlorothiazide 10 mg/kg/day 或 Hydrochlorothiazide 1–2 mg/kg/day - 4️⃣ Somatostatin analogue(Octreotide)
• Starting dose:5–10 μg/kg/day,最高 20 μg/kg/day,SC Q6H
⚠️ 小心 necrotizing enterocolitis(NEC)!
- 🔬 藥物(Diazoxide)無反應、考慮手術時,18F-DOPA PET/CT 是區分 focal 與 diffuse、並術前定位的首選影像:focal(父源 KATP 突變+病灶內母源等位基因缺失)可局部切除治癒;diffuse 藥物無效時多需近全(95–98%)胰切除。doi:10.1159/000531766
Sperling Pediatric Endocrinology, 6th ed., Chapter 7.
De Leon DD, Arnoux JB, Banerjee I, et al. International Guidelines for the Diagnosis and Management of Hyperinsulinism. Horm Res Paediatr. 2024;97(3):279–298. doi:10.1159/000531766. Epub 2023 Jul 14.
- 最常見:dumping syndrome(Nissen fundoplication / gastric bypass 後)
- 機轉:GLP-1 過度分泌 → 胰島素爆發
- 檢查:mixed meal test / OGTT / hyperglucidic breakfast test
非酮性低血糖(Hypoketotic hypoglycemia)
- AKT2 activating mutation(影響 insulin receptor)
- PI3K activating mutation(影響 insulin receptor)
- MORFAN 症候群(mental retardation、pre-/postnatal overgrowth、remarkable face、acanthosis nigricans;de novo AKT2)
- Abnormal IGF-II processing → NICTH(non-islet cell tumour hypoglycaemia):腫瘤產生大量未完全處理的高分子量 "big" pro-IGF-II,具類胰島素活性
人為低血糖(Factitious hypoglycemia)
- 來源:外源 insulin 或 sulfonylureas(刺激內源胰島素)
- 關鍵生化:外源 insulin → insulin 高但 C-peptide 正常(insulin/C-peptide ratio > 1)
反調節荷爾蒙衰竭(Failure of counter-regulatory hormone)
- 先天性 hypopituitarism 可致危及生命低血糖、鈉異常、休克、conjugated hyperbilirubinaemia、microphallus、生長遲滯
- Panhypopituitarism 低血糖發生率可達 20%,甚至猝死
- 治療:hydrocortisone + GH 補充(壓力時加量)
- 基因:POU1F1、PROP1、LHX3、LHX4、SOX3、OTX2、GLI2、HESX1;也見於 Bardet-Biedl、Prader-Willi
- 後天:顱咽管瘤最常見、放射、感染、外傷
- 症狀:疲倦、姿勢性頭暈、肌無力、體重↓、鹽渴、色素沉著
- 確診:低 cortisol + 顯著↑ ACTH(次發/三發性無皮膚變化)
- 分三類:dysgenesis / impaired steroidogenesis / destruction
- 1️⃣ Adrenal dysgenesis or hypoplasia
- 基因:NR5A1(SF1)、NR0B1(DAX-1, X-linked)、CDKN1C(IMAGe)、SAMD9(MIRAGE)
- SF-1 缺陷 → 腎上腺衰竭 + XY 性反轉
- DAX-1 → 合併 hypogonadotropic hypogonadism(可為 contiguous gene syndrome,合併 glycerol kinase deficiency、DMD)
- FGD(familial glucocorticoid deficiency)
- AAA / 4A 症候群(achalasia、alacrima、adrenal insufficiency、autonomic)
- 2️⃣ Impaired steroidogenesis
- CAH(21-OHD, CYP21A2)最常見(發生率 1/15,000;carrier 1/50)
- 完全缺陷 → salt-wasting(女性產前男性化、新生兒失鹽危象)
- 男嬰首發現線索:出生 1–8 週 collapse + 低血糖 + 低血壓 + 高血鉀
- 3️⃣ Adrenal destruction
- 自體免疫佔 ~50%(可為 polyglandular syndrome 一部分)
- Adrenoleukodystrophy(男性測 plasma VLCFA)
先天性代謝異常(Inborn errors of metabolism)
- 非內分泌領域,詳見教科書相關內容敘述。
Sperling Pediatric Endocrinology, 6th ed., Chapter 7.
Brook's Clinical Pediatric Endocrinology, 7th ed., Chapter 16.